咨詢熱線
17621170138
17621170138

除了熒光的光譜位置外,熒光量子產率(η fl;英文fluorescence quantum y field,QY)是熒光團的一個重要光學參數。除了消光系數的大小,當染料用作熒光標記時,量子產率的水平特別決定了獲得的信號強度:因此在英語用法中,消光系數和量子產率的乘積被稱為作為熒光團的“亮度"。
量子產率本質上取決于染料的分子結構,但也受到許多外部因素的影響。這些包括環境的溫度、粘度、極性和pH值。這種環境可以由溶劑分子和任何溶劑組成,也可以由例如偶聯的生物分子或細胞膜組成,熒光染料位于其附近。通過改變熒光,可以得出有關環境某些特性的結論:可以說,熒光染料充當分子探針。
熒光量子產率定義為以熒光形式發射的光子數(= 光量子)與樣品先前吸收的光子數之比:如果所有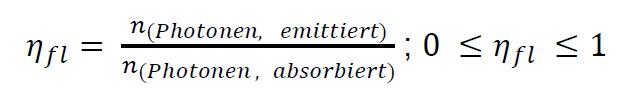
吸收的光子都以熒光形式發射,則量子產率為 1 或 100 % 獲得。然而,激發態的非輻射失活過程總是與發射競爭,因此部分吸收的能量以熱量的形式釋放到環境中。
正是這種現象被用于絕對判定通過“量熱"方法(例如熱開花方法)的量子產率。這需要相對復雜的測試設置以及對測量概念和評估的全面理論理解。
更簡單地說,熒光團(樣品)的未知量子產率可以通過將其與熒光光譜儀中標準或參考染料(參考)的已知量子產率進行比較來確定。這種所謂的相對確定可以通過不同的方式進行:
在單次測量中比較樣品與參考染料
在幾個單獨的測量中將樣品與幾種參考染料進行比較
將樣品與不同濃度的參考染料進行比較,并對獲得的測量值進行后續評估。
結果的統計準確性隨著進行的比較測量的次數而增加。
使用相對方法的理論先決條件是要比較的兩種溶液,樣品和參考,在激發波長下具有相同的吸收,因此吸收相同數量的光子。然后兩種溶液在相同條件下記錄的積分熒光光譜的商(IF = 熒光帶的面積)給出兩種染料的量子產率比,這樣就可以很容易地計算出未知的量子產率: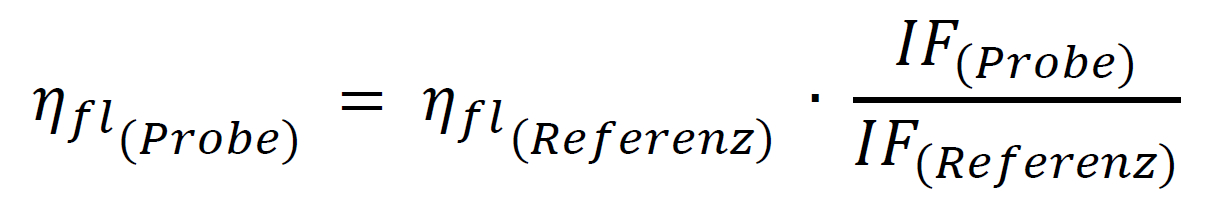
記錄樣品和參考的整個熒光光譜(積分熒光強度)的重要性通過以下示例用這種比較方法進行了說明:染料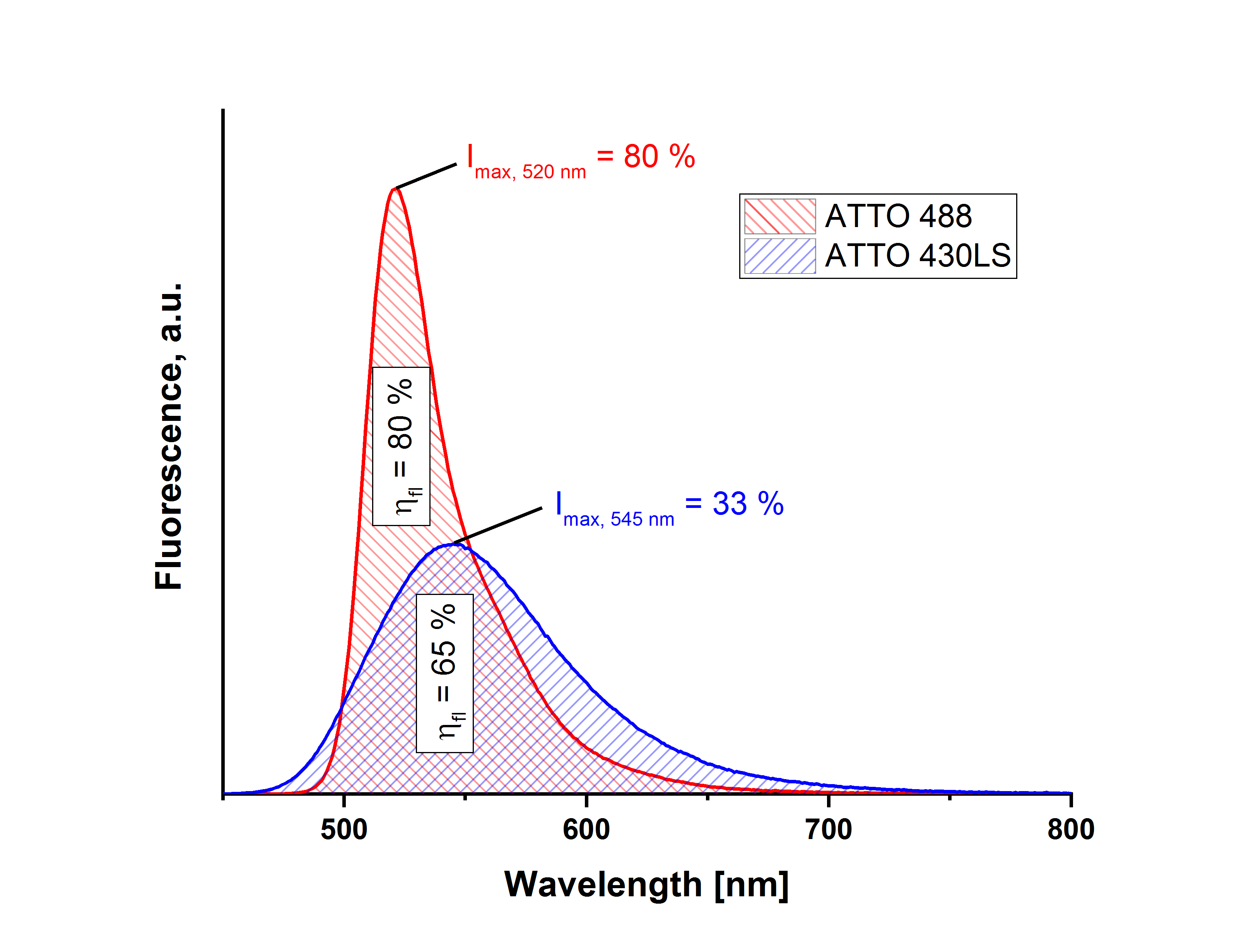
ATTO 488羧基和ATTO 430LS羧基在吸收光譜的交叉點被激發(相同的吸收) 和測量的熒光光譜。與標準品(羅丹明 6G)的比較測量導致ATTO 488羧基的熒光量子產率為 80%。使用此值,使用描述的相關方法獲得ATTO 430LS的 65% 的值羧基。另一方面,如果您只查看相應熒光最大值處的強度,您只能從ATTO 488羧基的已知 80% 中獲得 33% 的ATTO 430LS羧基。這種強烈的差異是由于熒光帶的寬度不同,即兩種染料的熒光光譜分布不同。 通過使用相同的測量參數,可以從設備端實現用于記錄樣品和參考熒光光譜的相同條件。這些包括光束路徑的幾何形狀(例如 90° 或正面布置)、檢測器放大(增益)、狹縫寬度(或帶通)和相同的激發波長。
例如,可以選擇樣品和參比的長波吸收帶的交點作為激發波長。然后,在染料條帶的相應最大值處的吸收可能略有不同。
使用示例 阿托 488 羧基和 阿托 532 羧基在各自的吸收最大值處的吸光度值略有不同,這一點變得很清楚:在記錄了兩種測量溶液的吸收光譜后,可以通過疊加兩種光譜來確定兩種溶液具有相同吸光度(此處為0.0183)的交點(此處為513.1 nm)。(對于較小的值,吸光度和吸光度相同。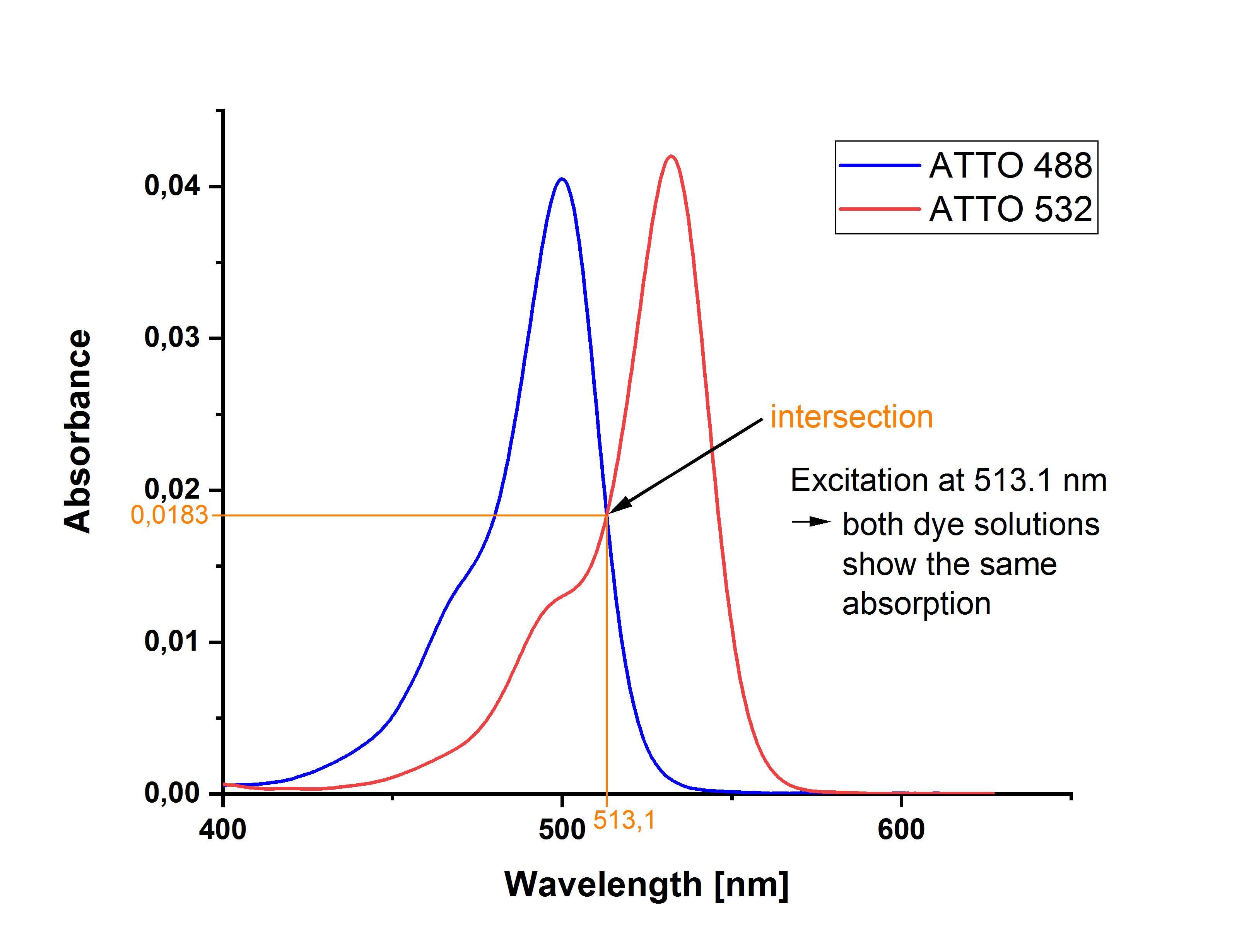
另一方面,如果決定在樣品和參考的吸收最大值處激發,則兩種溶液在各自波長下必須具有相同的吸收。在所示示例中,ATTO 488羧基可在 500 nm 激發,ATTO 532羧基可在 532 nm 激發;兩種溶液的吸光度均為 0.0420。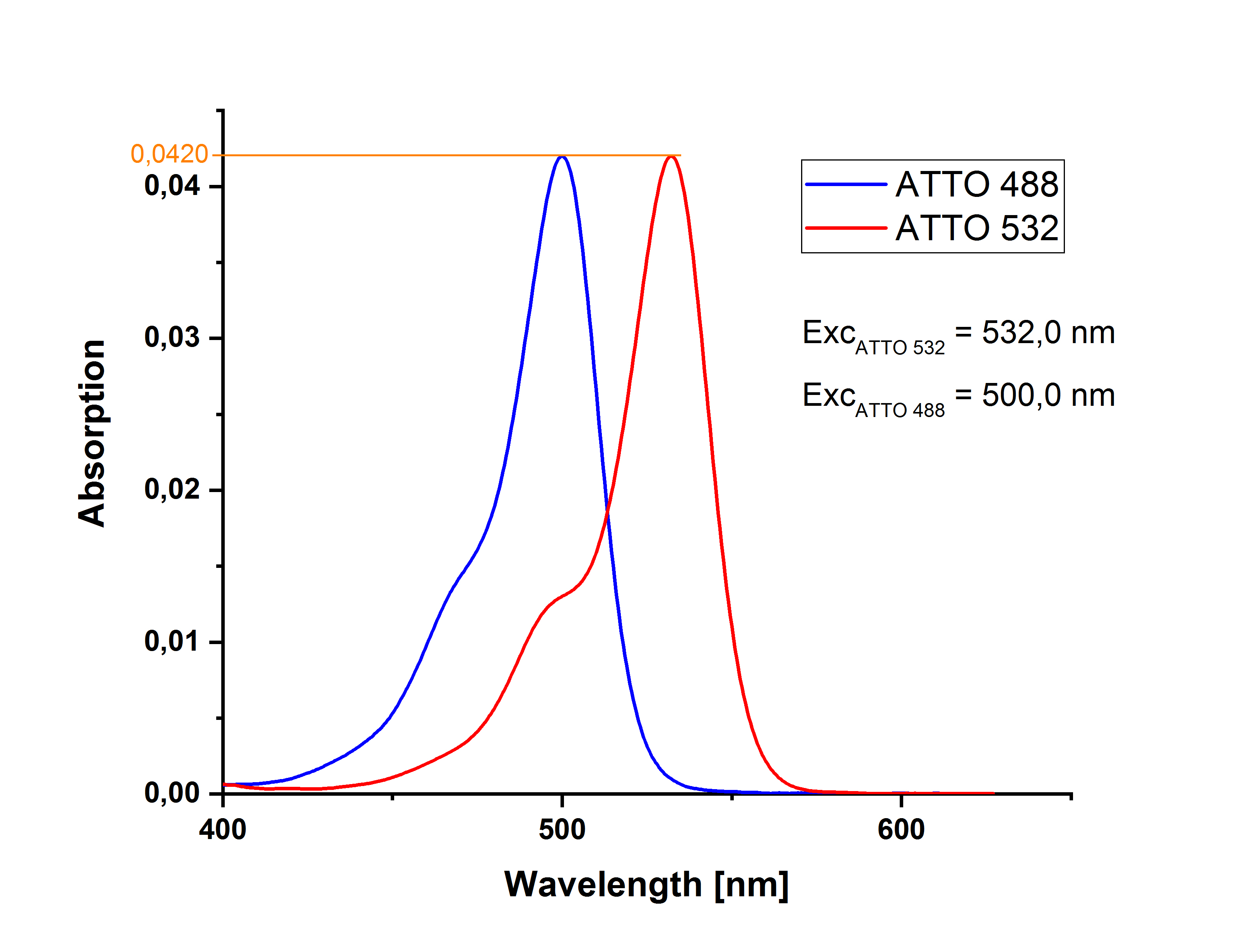
在這種情況下,必須在熒光測量中對兩個激發波長下的不同激發強度進行相應的校正。在現代熒光光譜儀中,這種與波長相關的強度差異由光電二極管確定為標準,同時還測量激發光源隨時間的強度波動。此校正必須在測量期間通過設備軟件激活,因為此后將無法再進行。
此外,所選測量范圍應覆蓋整個熒光光譜,即長波邊緣的測量強度應幾乎降至檢測器噪聲水平。
即使滿足所有這些要求,熒光量子產率的精確測定在實踐中也非常苛刻,需要做好充分的準備和仔細的測量。
關于量子產率測量和可能的誤差來源的說明
參比染料必須合適,并且必須足夠精確地知道其量子產率。例如,可以通過將其與另一個參考進行比較來確保這一點。
參比染料的選擇方式應使其能夠在與樣品相同的范圍內被吸收,即激發。理想情況下,兩個吸收光譜重疊,并且可以在熒光測量期間在兩個波段的交叉點激發。
必須保持所有玻璃器皿和比色皿絕對清潔。
使用的溶劑應具有“光譜學"規格,并應檢查其固有熒光。
例如,如果由于溶解度的差異而將不同的溶劑用于樣品和參比,則必須通過在計算中包括兩個折射率n來考慮這一點:
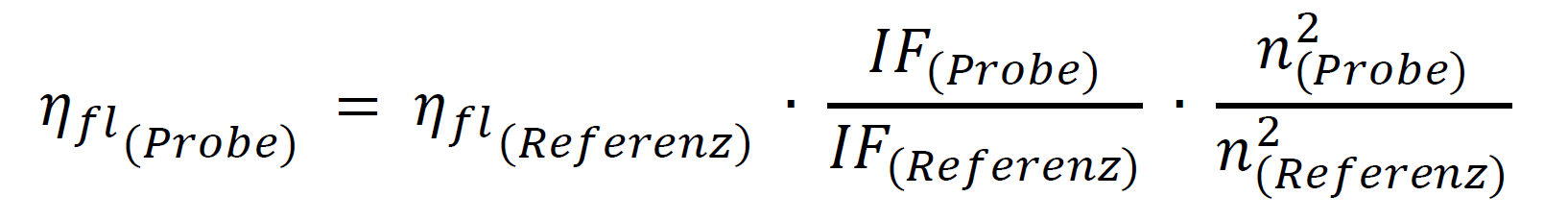
對于熒光測量,應使用光程長度為 10 mm 的標準熒光比色皿。
為了減少重吸收效應的發生,10mm電池中的吸光度不應超過0.05。
在較高濃度下,可能會發生所謂的內部濾光片效應,這極大地偽造了熒光測量:
一方面,激發光不再深入溶液,這可能導致比色皿中心的樣品激發減少。
另一方面,從那里發出的熒光在離開比色皿之前通過溶液時被光束路徑中的其他熒光團部分(重新)吸收。這導致熒光光譜在短波范圍內被切斷。
必須注意確保吸收測量的基線不會因未溶解顆粒或臟比色皿窗口的光散射而失真。
順便說一下,這種散射現象也會干擾熒光測量,因為散射光可以到達溶液中顆粒或比色皿表面上的檢測器。因此,在測量前應μ過濾所使用的溶劑和溶液,并用不起毛的布從外部擦拭比色皿窗口。注意:指紋!
熒光測量的測量參數(增益、狹縫寬度)必須適應發生的熒光強度,以便所使用的檢測器(例如光電倍增管)不會因過多的光而損壞。測量必須在探測器的線性范圍內進行,因為只有這樣,測量的光強度才會與照射的光強度成正比。
人們應該意識到溫度對測量結果的影響。
許多制造商現在都包含使用其熒光光譜儀測量熒光量子產率的精確說明和工作說明,也可以從相應網站下載。通常也可以在這里找到有關相應設備設置和測量參數的詳細信息和注意事項。正在努力開發熒光標準的方法和程序。我們給出的熒光光譜是用HORIBA Jobin Yvon的
Fluorolog 3熒光光譜儀測得的。為此,ATTO-染料在 22°C 下檢查水溶液(PBS,pH 7.4)中的相應羧基衍生物。測量是在標準的 90° 排列中進行的,具有水平的激發和發射極化。為了確定ATTO染料的熒光量子產率,將具有熒光量子產率的熒光團用作參考染料。取決于光譜范圍,例如B、使用羅丹明6G、羅丹明630等。
熒光光譜的一般信息
在吸收光譜儀的情況下,幾乎只使用雙光束裝置,其中樣品溶液和參比物質(例如帶有純溶劑的比色皿)“同時"掃描。結果,獲得的頻譜成為所謂的 設備特性 這是由于單色器(光柵、狹縫)和鏡子反射的不同透射率,除其他外,對于不同的偏振光和特定的檢測器特性。
由于熒光光譜中原則上不存在這種參考光束路徑,因此必須以不同的方式校正每個熒光光譜儀的器件特性,具體取決于光學元件和光束路徑的路徑,這些特性是不同的,以獲得樣品的“真實"熒光光譜。
在現代熒光光譜儀的情況下,控制和評估軟件通常包含所謂的 校正功能,可用于在測量過程中直接或通過用戶的后續指令校正被測設備光譜。此校正功能由制造商創建,例如,通過將相關設備測量的發射光譜與校準燈的實際發射光譜進行比較。
除此之外,在每次熒光測量中都應考慮另一種現象。這些是 熒光偏振: 只有當熒光團在激發態的生命周期內能在培養基中自由移動時,才能獲得非偏振熒光。在這種情況下,水平和垂直偏振熒光的比例是相同的。
自由遷移率受溶劑的溫度相關粘度和分子體積的影響。
在丙酮、甲醇、乙醇和水等低粘度溶劑中,這種極化效應通常只在室溫測量中壽命在納秒范圍內的小型有機熒光團中起次要作用。
但是,如果樣品和/或參比的熒光由于分子尺寸和/或熒光壽命的強烈不同而不再非偏振甚至不同的偏振,則可能導致測量的熒光光譜偽造,從而導致錯誤計算的量子產率。
為了確定熒光基團溶液的熒光偏振,有一種相對簡單的方法,可以在J. R. Lakowicz中找到其理論推導和解釋。
研究與大分子(聚合物、蛋白質、DNA等)偶聯的熒光標記物的熒光偏振,其自由遷移率在其激發態的生命周期內受到限制甚至受到抑制,可以與FRET技術相結合,為分子結構(幾何形狀,距離,方向)和相關動態現象提供重要的見解。
 掃一掃,關注微信
掃一掃,關注微信 當前位置:
當前位置: